El cromosoma 22 es uno de los cromosomas más conocidos debido a los patrones de síntomas que pueden producir, esto es debido tanto la falta de material genético, como una dosis genética extra en el mismo. La falta o exceso de material genético del cromosoma 22 es debido a los diferentes reordenamientos que éste puede producir y que se manifiestan clínicamente de modo distinto.
Los rasgos clínicos descritos a continuación, se describen según el número de casos presentados por la literatura científica. Pero debemos recordar que existen otros factores, además de la genética, por los que la sintomatología clínica se puede presentar de formas más leves o agudas.
Algunos de estos rasgos son accionables y lo que nos permite la Genética en nuestros días es tener diagnósticos anticipados para poder planificar clínicamente la vida con estos síndromes.
Los síndromes más conocidos que se dan en la región cromosómica 22q son:
SÍNDROME DE DELECCIÓN 22q11
El síndrome 22q11.2 es una anomalía cromosómica que causa un cuadro clínico con malformaciones congénitas cuyos rasgos característicos incluyen defectos cardíacos, anomalías del paladar, dismorfismo facial, retraso en el desarrollo e inmunodeficiencia.
Se estima una incidencia global de 1/2.000-1/4.000 nacidos vivos.
En la mayoría de los casos su origen es debida a una delección de aproximadamente 3 Megabases (millones de pares de bases) de la región cromosómica 22q11.2. Esta región cromosómica está rodeada de regiones cromosómicas que se repiten en bajo número de copias; suele tener su origen durante la espermiogénesis u ovogénesis (proceso de formación de los gametos parentales).
La delección que puede presentar un/a paciente puede ser de tamaño variable, a veces es menor de 3Mb en el 15% de los casos., mientras otras veces pueden ser delecciones aípicas que comprenden dicha región.
Lo que sí sabemos es que en el 22q11.2 se encuentra una región cromosómica crítica que cuando está deleccionada produce un fenotípico característico. Esta región crítica se conoce como ¨región crítica DiGeorge¨, por ello este síndrome también es conocido como Síndrome de DiGeorge. Dentro de esta región crítica se encuentra el gen TBX1 que está implicado en el desarrollo cardíaco, las paratiroides, el timo y la estructura facial.
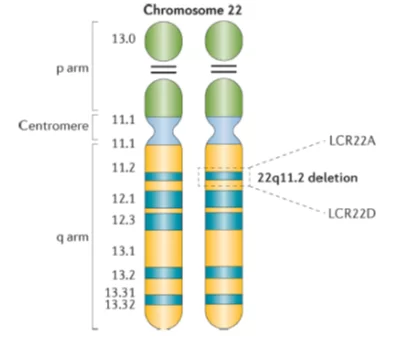
Algunos de los rasgos físicos más frecuente de este síndrome son: Anomalías cardiacas, funcionamiento deficiente del sistema inmunitario, hendidura del paladar, complicaciones con bajos niveles de calcio en sangre, retraso en el desarrollo y problemas tanto de conducta como emocionales.
SÍNDROME DE DUPLICACIÓN 22Q11
Una duplicación 22q11.2 es una variación genética en el cual hay una copia adicional de una pequeña parte del cromosoma 22. La duplicación se encuentra cerca de la mitad del cromosoma, en la banda cromosómica q11.2. Debido a que la parte adicional es muy pequeña se le llama microduplicación.
Las características de las personas con duplicación 22q11.2 varían enormemente, incluso entre los miembros de una misma familia. Los portadores de este síndrome pueden presentar retraso en el desarrollo, discapacidad intelectual, crecimiento lento que conduce a baja estatura y tono muscular débil (hipotonía). Algunas personas con la duplicación no tienen dificultades físicas ni discapacidades intelectuales, por lo que no podemos estar del todo seguros de la causa y el efecto de esta microduplicación. En este último caso hablamos de duplicación ¨silenciosa¨, y algunos padres y hermanos/as de los pacientes que sufren este síndrome son portadores de la misma duplicación 22q11.2 pero sin síntomas o signos destacables.
El tamaño de la región cromosómica que se duplica en este síndrome es de unos 1.5 Megabases, pero existen pacientes con duplicaciones más pequeñas. Estas personas parecen verse afectadas de maneras muy diversas. Dependiendo de los genes que se dupliquen presentarán signos diferentes.
Los principales signos físicos de esta duplicación son: Anomalías cardiacas, insuficiencia velofaríngea (hendidura del paladar), pérdida de audición, retraso del crecimiento, necesidad de apoyo en el aprendizaje, problemas de comportamiento y características faciales inusuales.
SÍNDROME DE DELECCIÓN DISTAL 22Q11.2
Los rasgos del síndrome de delcción distal 22q11.2 son debidos a la pérdida de unos genes situados en esta región. Se trata de uan delección pequeña, donde la mayoría de pacientes tienen pérdidas cromosómicas de 0,4 a 2,1 Mb (400.000 a 2 Millones de bases).
Los genes implicados en esta delección no están descritos del todo. Se han propuesto que los genes CRKL y MAPK1 juegan un papel en las anomalías cardiacas que son comunes para este síndrome. El gen MAPK1 parece estar asociado a la prematuridad y el retraso de crecimiento intrauterino durante el embarazo, ya que parece estar asociado en el crecimiento de la placenta. También parece estar relacionado con el comportamiento, según algunas publicaciones en ratones como modelo animal. En delecciones muy distales que incluyen al gen SMARCB1 se asocian con un riesgo aumentado de tumores rabdoides.
Algunos de los rasgos más comunes que presentan las personas con esta delección son:
Retraso del lenguaje (problemas en la articulación),retraso en el crecimiento, problemas cardiacos, dificultades en la concentración, rasgos faciales sutiles, necesidad de apoyo en el aprendizaje, discapacidad intelectual de leve a moderada.
SÍNDROME DE EMANUEL
El síndrome de Emanuel lleva el nombre de la Dra. Beverly Emanuel, una experta en citogenética de Filadefia. En la literatura científica este síndrome se cita como Síndrome de derivado 22 o trisomía parcial 11;22.
Las personas que son portadoras de este síndrome tienen 47 cromosomas, en vez de 46.
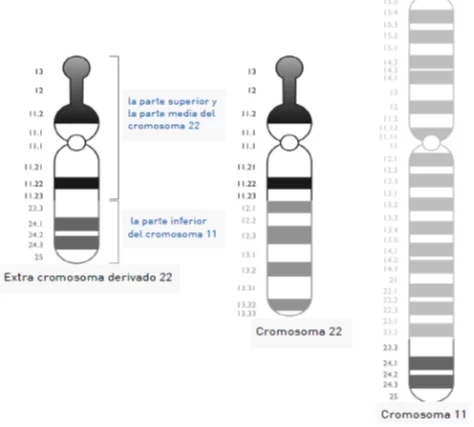
El cromosoma extra encontrado en este síndrome se compone de una parte de cromosoma 22 y otra parte del cromosoma 11. Este cromosoma derivado extra se hereda de un padre o una madre con lo que llamamos una traslocación entre los cromosomas 11 y 22.
Es decir, el padre o la madre son portadores sin síntomas donde se han intercambiado los cromosomas 11 y 22, pero tienen 46 cromosomas.
Normalmente este cromosoma 22 derivado extra proviene de la madre.
La incidencia de este síndrome es desconocida, pero sabemos que es un síndrome poco habitual.
Algunos signos que presentan los portadores de este síndrome son: Hipotonía, paladar hendido, defectos cardiacos, malformaciones en los riñones, pérdida de audición de moderada a profunda, infecciones recurrentes destacando el oído, desarrollo leve del lenguaje, convulsiones, diversas discapacidades físicas e intelectuales.
SINDROME DEL OJO DE GATO
El síndrome ojo de gato o ¨Cat Eye Syndrome¨ en inglés, es un síndrome poco frecuente con rasgos físicos variados. Uno de los signos físicos más característicos es una anomalía del iris llamada ¨coloboma del iris¨. Los tres signos visibles principales de este síndrome son: las anomalías preauriculares, la atresia anal y el coloboma del iris; pero ninguna de las tres se encuentra de manera constante.
Tiene una prevalencia al nacimiento estimada en 1/50.000 a 1/150.000, afectando de forma similar a hombre y mujeres.
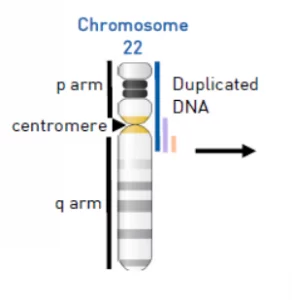
La causa citogenética de este síndrome es una copia extra de la región de 22q11 hasta el telómero 22pter del cromosoma 22. En un tercio de los casos este cromosoma extra aparece en mosaico, es decir, que no aparece en todas las células, solo aparece en un porcentaje de células.
Al analizar el cariotipo aparece en forma de cromosoma marcador o supernumerario, de modo que contaríamos 47 cromosomas en vez de 46.
Otros rasgos físicos que podemos físicos que podemos encontrar en este síndrome son: rasgos faciales con fisuras palpebrales, labio leporino o paladar hendido, el coloboma del ojo puede afectar al iris, la coroides y/o la retina. Defectos cardiacos congénitos, anomalías de riñón, anomalías óseas. Hernias, criptorquídias e hipospadias en hombres. La mayoría presentan discapacidad intelectual leve. En algunos casos se desarrolla talla baja asociada con deficiencia de la hormona de crecimiento.
SINDROME DEL 22 EN ANILLO
El Síndrome del 22q en anillo es un síndrome poco frecuente que se caracteriza por encontrar al cromosoma 22 en forma circular en vez de en forma elongada, tal como se encuentran los cromosomas habitualmente.
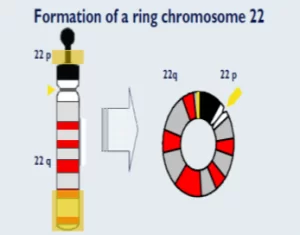
Los cromosomas en anillo llevan a una pérdida de material genético en la zona terminal del cromosoma. Normalmente para la formación de este tipo de cromosoma hay una rotura de las partes terminales que acaban perdiéndose. Dependiendo de la región cromosómica perdida, se presentarán una serie de rasgos físicos. El gen SHANK3 es uno de los genes situados en la parte terminal del cromosoma 22q, y cuando se pierde este gen podemos tener una sintomatología típica del cromosoma 22 en anillo.
Hay una gran variabilidad en los síntomas, aunque en una mayoría se presentan como leves. Algunos de ellos son: Anomalías cardiacas leves, convulsiones, infecciones respiratorias, problemas de sueño y problemas de alimentación por reflujo, dificultades de aprendizaje leves e hiperactividad.
Este síndrome tiene sintomatología muy similar al Síndrome Phelan-McDermid que se produce por una delección de la región q terminal 22q13.
Mercedes Navarro.
ASESORAMIENTO GENÉTICO.
Dirª. Mercedes Navarro, M.Sc.
Preguntas frecuentes
- ¿Qué es el síndrome de delección 22q11?
- El síndrome 22q11.2 es una anomalía cromosómica que causa un cuadro clínico con malformaciones congénitas, incluyendo defectos cardíacos, anomalías del paladar, dismorfismo facial, retraso en el desarrollo e inmunodeficiencia.
- ¿Cuál es la causa del síndrome de delección 22q11?
- En la mayoría de los casos, el síndrome de delección 22q11.2 se debe a una delección de aproximadamente 3 Megabases de la región cromosómica 22q11.2. Esta delección ocurre durante la espermiogénesis u ovogénesis.
- ¿Qué es el síndrome de duplicación 22q11?
- El síndrome de duplicación 22q11.2 es una variación genética en la que hay una copia adicional de una pequeña parte del cromosoma 22. Las características de las personas con este síndrome varían enormemente, incluso entre miembros de una misma familia.
- ¿Cuáles son los signos más comunes del síndrome de duplicación 22q11?
- Los signos más comunes incluyen anomalías cardiacas, insuficiencia velofaríngea, pérdida de audición, retraso del crecimiento, necesidad de apoyo en el aprendizaje, problemas de comportamiento y características faciales inusuales.
- ¿Qué es el síndrome de delección distal 22q11.2?
- El síndrome de delección distal 22q11.2 es una condición genética causada por la pérdida de genes en esta región. Los genes CRKL y MAPK1 se han asociado con las anomalías cardiacas y el retraso de crecimiento intrauterino.
- ¿Qué síntomas presenta el síndrome de delección distal 22q11.2?
- Los síntomas incluyen retraso del lenguaje, problemas cardiacos, dificultades en la concentración, rasgos faciales sutiles, necesidad de apoyo en el aprendizaje y discapacidad intelectual de leve a moderada.
- ¿Qué es el síndrome de Emanuel?
- El síndrome de Emanuel, o síndrome de derivado 22, es una condición genética en la que los individuos tienen 47 cromosomas en lugar de 46, con un cromosoma extra compuesto de partes de los cromosomas 11 y 22.
- ¿Cuáles son los signos del síndrome de Emanuel?
- Los signos incluyen hipotonía, paladar hendido, defectos cardiacos, malformaciones en los riñones, pérdida de audición, infecciones recurrentes, desarrollo leve del lenguaje y diversas discapacidades físicas e intelectuales.
- ¿Qué es el síndrome del ojo de gato?
- El síndrome del ojo de gato es una condición genética rara caracterizada por una copia extra de la región 22q11 hasta el telómero 22pter del cromosoma 22. Los tres signos visibles principales son anomalías preauriculares, atresia anal y coloboma del iris.
- ¿Cuáles son otros rasgos físicos del síndrome del ojo de gato?
- Otros rasgos incluyen fisuras palpebrales, labio leporino, paladar hendido, defectos cardiacos congénitos, anomalías de riñón, anomalías óseas, hernias y discapacidad intelectual leve.
- ¿Qué es el síndrome del 22 en anillo?
- El síndrome del 22 en anillo es una condición genética en la que el cromosoma 22 está en forma circular en lugar de elongada, lo que lleva a la pérdida de material genético en la zona terminal del cromosoma.
- ¿Cuáles son los síntomas del síndrome del 22 en anillo?
- Los síntomas incluyen anomalías cardiacas leves, convulsiones, infecciones respiratorias, problemas de sueño, problemas de alimentación por reflujo, dificultades de aprendizaje leves e hiperactividad.